| |
| |
| |
| |
|
20220919M: Fluorescence intensity decreases with increasing temperature. As a "rule of thumb", you can assume 1% decrease in intensity for every 1 degree Celsius (or Kelvin) increase in temmperature. For example, if 22C is 100% then 37 C may be 85% (100 - 15).
Some fluorophores have a larger "delta", some up to 5% decrease per 1 degree C increase -- these are typically "molecular rotors" that respond to viscosity changes (viscosity at the scal of the molecule).
Agarwal R 2008 Temperature sensitivity and fluorescence detection. J. Sep. Sci. 31: 128 – 132. ... this publication also found that their Mercury arc lamp illuminator (directly coupled to their fluorimeter) increased the temperature at the cuvette to be measrable. Most modern microscopes have liquid light guide (ex. FISHscope, our Zeiss inverted) or optical fiber (confocal microscopes) between the light asource and microscope.
|
|
20220520F - one of our users, Olga K, likes FluorSave [may sometimes be misspelt FluoroSave) from MilliporeSigma. Questions came up of refractive index. Olga likes it because solifdifies (in slide coverglas mount) in ~1 hour. GM note: so what is it shrinks 1 week later? This could be considered a good thing, since smaller volume = faster imaging (the opposite of goal of Ed Boyden's ExM Expansion Microscopy!). As long as you know what is happening to your specimen. I also note that "R.I. goes up after drying" (solidifying) may put the specimen/mount at extremely close to the refractive index of our Olympus Silicone Oil (1.406), enabling better confocal image quality (our FV3000RS has 30x/1.05, 40x/1.25, 100x/1.35 NA S.I lenses).
Confocal listserv message:
Fluorsave (Calbiochem à MilliporeSigma)
1.358 wet
R.I. goes up after drying
Wes Wallace - I personally recommend Fluorsave from Calbiochem. It does not contain glycerol, and its antifade properties seem to be quite strong. However, it does have the disadvantage described in one of the earlier posts, namely after about a week it seems to shrink and distort the tissue. (confocal listserv Mon, 11 Feb 2002 13:12:10 -0500 )
https://lists.umn.edu/cgi-bin/wa?A2=ind0202&L=CONFOCALMICROSCOPY&P=R4934&X=2995B898A545DAF92D&Y
|
|
Rea 20220301 Mike Rea quoting Mark Twain cant trust your eyes when your imagination is out of focus - science conversation between rigor and imagination dialogue not monologue
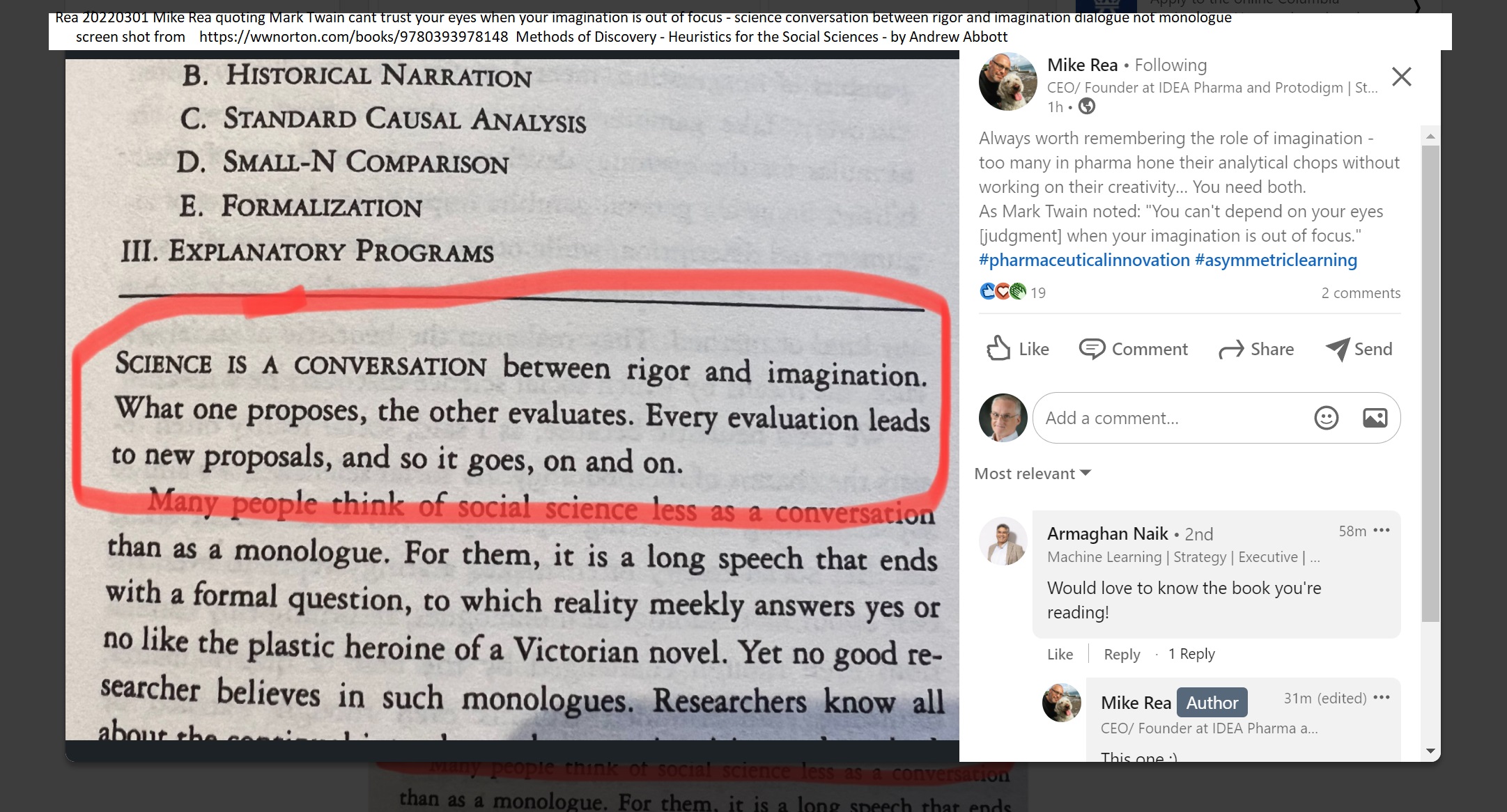
screen shot on left from:
Methods of Discovery - Heuristics for the Social Sciences
by Andrew Abbott (Author, University of Chicago)
https://wwnorton.com/books/9780393978148
|
|
Clever method to calibrate fluorescence intensity to number of molecules (note: paper also used IRM, interference reflection contrast microscopy, one of my favorite imaging methods)
Mechanisms of frustrated phagocytic spreading of human neutrophils on antibody-coated surfaces
Emmet Francis, Hugh Xiao, Lay Heng Teng, Volkmar Heinrich
doi: https://doi.org/10.1101/2022.02.18.481104 https://www.biorxiv.org/content/10.1101/2022.02.18.481104v1 Posted February 19, 2022.
This standard consisted of batches of microspheres pre-functionalized with known numbers of antibody-binding sites (QSC Kit; Bangs Laboratories, Fishers, IN) that we saturated with the same secondary antibody as used to label the coverslips (Fig. 1B). We analyzed Z-stacks of confocal images of these beads to determine the intensity of the fluorescent layer at the underside of each bead local to the center of the bead image (Materials and methods; Fig. S1). This analysis provided a calibration curve relating the fluorescence intensity of an essentially flat layer of secondary antibody to the known surface density of rabbit IgG (Fig. 1B). The calibration curve then allowed us to convert measurements of the mean fluorescence intensity of confocal images of labeled coverslips (Fig. 1A) to the density of rabbit IgG deposited on these coverslips. We note that the fluorescence intensity of the most densely coated coverslips lay outside the intensity range of the bead standards; in this case, our analysis involved an extrapolation of the calibration curve.
|
|
NIH data sharing policy requirement starts January 25, 2022
1. Nature news story 2/2022.
2. headline of NIH notice (posted October 2020)
3. Existing JHU CRAEDL https://craedl.org data repository.
4. New (3/11/2022) information on newly launched Johns Hopkins University School of Medicine Archive.
***
NIH issues a seismic mandate: share data publicly
The data-sharing policy could set a global standard for biomedical research, scientists say, but they have questions about logistics and equity.
Max Kozlov
https://www.nature.com/articles/d41586-022-00402-1
In January 2023, the US National Institutes of Health (NIH) will begin requiring most of the 300,000 researchers and 2,500 institutions it funds annually to include a data-management plan in their grant applications — and to eventually make their data publicly available.
Researchers who spoke to Nature largely applaud the open-science principles underlying the policy — and the global example it sets. But some have concerns about the logistical challenges that researchers and their institutions will face in complying with it. Namely, they worry that the policy might exacerbate existing inequities in the science-funding landscape and could be a burden for early-career scientists, who do the lion’s share of data collection and are already stretched thin.
The mandate, in part, aims to tackle the reproducibility crisis in scientific research. Last year, a US$2-million, eight-year attempt to replicate influential cancer studies found that fewer than half of the assessed experiments stood up to scrutiny. Efforts to tally the cost of irreproducible research in the United States have found that $10 billion to $50 billion is spent on studies that use deficient methods, a cost that is mostly fronted by public funding agencies.
Irreproducible studies not only waste taxpayers’ money, says Lyric Jorgenson, the acting associate director for science policy at the NIH, but also undermine public trust in science. “We want to make sure that we’re making good on the nation’s investment and fostering transparency and accountability in research,” she says.
Joseph Ross, a health-policy researcher at Yale School of Medicine in New Haven, Connecticut, says the mandate’s effects will be felt far beyond US borders because the NIH is the world’s largest public funder of biomedical research. Ensuring that the policy sets the right tone is important, Ross says, because it will signal to scientists all over the world how biomedical research should be done.
A seismic shift
Under the new policy, which will go into effect on 25 January, all NIH grant applications for projects that collect scientific data must include a ‘data management and sharing’ (DMS) plan that contains details about the software or tools needed to analyse the data, when and where the raw data will be published and any special considerations for accessing or distributing that data.
Such a seismic shift in practice has left some researchers worried about the amount of work that the mandate will require when it becomes effective.
Jenna Guthmiller, an immunologist at the University of Chicago in Illinois, can attest that more work will probably be required. She is one of a handful of researchers funded through a US National Institute of Allergy and Infectious Diseases (NIAID) programme that has enacted a policy similar to the NIH-wide plan, she says. For Guthmiller, that meant tracking down information on long-gone reagents and experimental conditions for a project that’s been running for four years. That took 15 hours, she says, “and I was fortunate enough to work with a data manager”.
Because the vast majority of laboratories and institutions don’t have data managers who organize and curate data, the policy — although well-intentioned — will probably put a heavy burden on trainees and early-career principal investigators, says Lynda Coughlan, a vaccinologist at the University of Maryland School of Medicine in Baltimore, who has been leading a research team for fewer than two years and is worried about what the policy will mean for her.
Jorgenson says that, although the policy might require researchers to spend extra time organizing their data, it’s an essential part of conducting research, and the potential long-term boost in public trust for science will justify the extra effort.
Others worry that data-management activities will further sap funds from under-resourced labs. Although the policy outlines certain fees that researchers can add to their proposed budgets to offset the costs of compliance with the mandate, it doesn’t specify what criteria the NIH will use to grant these requests.
For the policy to be successful, Ross says that the NIH needs to be clear about how it will award these resources — especially to early-career researchers and to underfunded institutions — so as not to exacerbate existing inequities in the research community.
Jorgenson responds that the agency is evaluating the costs of compliance and hopes to prepare more guidance and information.
Potential pitfalls
As part of the data-sharing policy, when a research project is complete or when its grant expires — whichever comes first — NIH programme officers will review the DMS plan to ensure that researchers have adhered to it. At that time, the policy stipulates that researchers must share any ‘scientific data’ needed to “validate and replicate research findings, regardless of whether the data are used to support scholarly publications” — although it makes an exception in cases where data sharing would pose a significant legal, ethical or technical burden. The NIH recommends that this data be shared only in a reputable repository; ultimately, researchers will decide where to upload the information.
The broad term ‘scientific data’ has left some researchers confused about exactly what information they’ll be required to share. It’s hard to predict which data might be useful for other researchers, or whether that data will ever be accessed by anyone, Coughlan says.
In response to an early draft of the policy, the American Association for Universities, an organization based in Washington DC that represents 66 universities, wrote in 2020 that the NIH’s definition of scientific data needed to be narrowed, and suggested that the agency limit it to include only data underlying scholarly publications.
Jorgenson says that data collected when experiments don’t work — and therefore that are not in publications — are just as important to communicate, because they include information that could help other researchers understand the full context of an experiment's success. The ambiguity in the policy offers researchers flexibility in determining which data are truly necessary to reproduce research findings, she says.
Brian Nosek, executive director of the Center for Open Science, based in Charlottesville, Virginia, points out that it will be a major challenge for the NIH to ensure that all relevant data have been shared at the conclusion of a project. Although the policy is an “important milestone of maturing the open-science movement beyond just thinking about open access”, Nosek worries that some applicants might not take it seriously if there are no consequences for non-compliance. Jorgenson responds that if the policy is not followed, future funding awards for researchers or institutions could be jeopardized.
Despite its potential pitfalls, Ross thinks that the policy will have a ripple effect that will persuade smaller funding agencies and industry to adopt similar changes. “This policy establishes what people expect from clinical research,” he says. “It’s essentially saying the culture of research needs to change.”
doi: https://doi.org/10.1038/d41586-022-00402-1
UPDATES & CORRECTIONS
Correction 16 February 2022: In an earlier version of this story, Lyric Jorgenson's surname was spelled incorrectly.
NIH Policy Notice 'headline' and JHU plan (brief)
|
https://grants.nih.gov/grants/guide/notice-files/NOT-OD-21-013.html
Final NIH Policy for Data Management and Sharing
Notice Number: NOT-OD-21-013
Release Date:October 29, 2020
Effective Date: January 25, 2023
Summary
The National Institutes of Health (NIH) is issuing this final NIH Policy for Data Management and Sharing (DMS Policy) to promote the management and sharing of scientific data generated from NIH-funded or conducted research. This Policy establishes the requirements of submission of Data Management and Sharing Plans (hereinafter Plans) and compliance with NIH Institute, Center, or Office (ICO)-approved Plans. It also emphasizes the importance of good data management practices and establishes the expectation for maximizing the appropriate sharing of scientific data generated from NIH-funded or conducted research, with justified limitations or exceptions. This Policy applies to research funded or conducted by NIH that results in the generation of scientific data.
* see https://grants.nih.gov/grants/guide/notice-files/NOT-OD-21-013.html for full notice.
|
|
JHU has had CRAEDL https://craedl.org as a open repository for data associated with JHU publications.
Craedl—the Collaborative Research Administration Environment and Data Library—is designed to support academic research by integrating cloud data storage, sharing, and discovery; research group project management; and institutional research administration. We provide secure, scalable research data management resources that allow academic research teams to focus on their core research so they can increase their productivity, improve their organization, and advance the state of their art.
|
|
JHU SOM Archive -- 3/11/2022 event to explain to SOM faculty, staff, students (online event that SOM'ers can find online and register for)
JHU Research Integrity Colloquium: Where are the Doggone Data for Figure 2? Introducing the SOM Archive
Description: Please join the Office of Research Integrity in welcoming Drs. Antony Rosen and Stuart Ray and David Kennedy, the Associate Director for Digital Systems and Services for Welch Medical Library for an introduction and discussion of the newly launched Johns Hopkins University School of Medicine Archive.
This conversation will feature a preview of the new Archive, place the SOM Archive in context together with other data management tools available, and allow you an opportunity to ask questions about the new tool.
|
|
|
Protecting objective lenses advice from confocal listserv ... gm summary: (i) lots of hands on training sessions for each user, (ii) find specimen focus with transmitted light, (iii) charge ~$1200 for each new user training [per instrument], partly to discourage non-serious PI's and their users.
Subject: Re: Protecting objectives
Date: Fri, 11 Feb 2022 13:46:24 +0000
From: Sylvie Le Guyader
To: CONFOCALMICROSCOPY@LISTS.UMN.EDU
Hello everyone
We have in place several measures that help greatly in the matter of damaged objectives:
1. Our training is spanned over several days (3 to 5) so we get at least 3 to 5 chances to input our routines into users heads and check that they follow them. Our routines therefore become users' routine due to the repetition.
2. To protect the objectives that are NOT in use and may bump into the stage as mentioned by someone else, leading to someone damaging the objective without even knowing it, we have capped the z drive of all our systems (Nikon) to a height safe for the other objective. At that height the objective in use may still be damaged but the others are safe. The great advantage of this is that we easily spot people who change samples and start using for example a multiwell plate with a too high skirt (btw we recommend to our users to use plates from Zell Kontakt and the reasons are here<https://microscopykarolinska.se/2017/08/25/glass-bottom-multiwell-plates/> if you are interested). They cannot focus so they come and ask us why then they get through the painful process of having to ask us to remove all the objectives not in use every time they use the microscope. They usually change plates very quickly.
3. Our software is set so that changing objectives always brings the objective turret to its lowest position first.
4. Our start up procedure is that everything is always restarted between 2 users even if the next user is standing by the microscope. For the reason written in point#1, I am very confident that all our users follow this procedure.
5. We train our users not to find their samples with fluorescence as the depth of field is much shallower than with transmitted light. Again see point#1 why they never do otherwise. We absolutely never look in the eyepiece at fluorescence. Our newer systems do not even offer this option and no user has ever noticed it. You are welcome to have a look at the technique we use to focus on the sample
https://www.youtube.com/watch?v=_R7BSM1krZQ
which works for ANY microscope with an eyepiece and a transmitted light source. Feel free to share/use the video.
6. We acquire a PSF every 2 months to check the objectives and as mentioned elsewhere, mostly identify problems before the users report them.
7. I personally enjoy creating a mutual trust relationship with users. I do not think users are evil or careless. But they are certainly stressed (as a biologist, been there done that). My experience with broken objectives is more with the objectives that are not in use. In 2017, we had 3 60x oil objectives damaged! It had happened once before in 10 years. It is likely that one user was using plates with a high skirt. At the end of 2017 we implemented point #2, we have had no objective damage since.
8. Our training costs 12,000 sek so we have 0 tourist and I see no reason why it should be otherwise. ��
Med vänlig hälsning / Best regards
Sylvie
Sylvie Le Guyader, PhD
Live Cell Imaging Facility Manager
Karolinska Institutet- Bionut Dpt
Hälsovägen 7C,
14157 Huddinge, Sweden
***
GM notes:
* SEK kroner 1.00 = US $0.1076 on 20220216W https://www.msn.com/en-us/money/tools/currencyconverter?duration=1D
so 12,000 sek is ~$1200.
* If we routinely specified 5 two hour training sessions per new user per confocal microscope, at "training rate" of $127/hr, thuis would be $1,270, very comparable to Sylvie's rate at KI. This might be good for a completely new to research microscopes user (ex. new graduate student or a postdoc moving from a non-microscope field). For an "experienced microscopist" -- if they had developed excellent work habits before startign to use our core, even 3 sessions would probably be too many. the real challenge is users who think they know what they are doing but have developed bad habits and do not learn new good habits.
|
|
More options to clean objective lenses -- from my MPMicro (Multi-Probe Microscopy) document pages 803-809 (2011 version, more content in same section) download at https://works.bepress.com/gmcnamara/2
• Rudi Rottenfusser (the resident Zeiss representative at Marine Biological Laboratory, Woods Hole, MA)
recommends for many types of dirt the use of “Sparkle” followed by blowing off residual Sparkle with a hand
held puffer (“squeeze-bulb”, a.k.a. “Dust Bulb” from ThermoOriel). Sparkle is apparently available by the gallon
at stores in Chicago (the most important point is that Sparkle is a non-ammonia glass cleaner that does not leave
a residue).
• One “recipe” I recall from Dr. Colin Izzard at a Woods Hole course is to make a 1:1 mixture of distilled water:ethyl alcohol (95% or absolute) and then mix it fresh with ether resulting in a 1:1:1 mix. The ether is flammable and volatile so be careful! {GM 20220216W ratios are clearly 1:1:2}
• We have also seen in print the use of 1:1 ether:ethanol (i.e. Richards and Richards, 1998). Like xylene, ether is a potential hazard in the lab, and should never be handled in rooms with flames.
• McBain Instruments (S. California Leica dealer) recommends Sparkle or methyl ethyl ketone (MEK), sparingly, on a cotton tip, to clean aqueous or organic build up on objective lenses. {gm note: MEK dissolves the anti-reflection coating from the front of the lens, so no longer recommended for normal use - may be useful sparingly to clean off 'nasty stuff' ... probably the dH2O : EtOH : Ether above should be considered first}.
• If you are unsure of how to remove the Sparkle or MEK or other solvent, please get advice from a professional (i.e. a microscope company or dealer microscope service person), or arrange for a professional to clean the equipment (it’s not that hard, but at several $K to replace or repair a damaged objective lens, you may want to avoid self-inflicted wounds to your budget).
• MEK, and especially solvents like ether, should be stored and used correctly. These both evaporate very quickly, so close the container tight as soon as you’ve removed enough solvent to do the job. Ether, especially, should never be used near a flame. See also the box below on handling solvents and in not applying so much that the optical cement dissolves.
MEK proved useful in un-sticking a Leica microscope condenser cap that would not unscrew. Following the advice of Adrian Powell, McBain Instruments, I loaded a small quantity of MEK into a 1 ml syringe, and used the needle to carefully position the MEK near the cap’s threads. After two applications, I was able to unscrew the cap. Note: the MEK also dissolved off the red ink Leica writing on the side of the cap.
• See also Chris Souwand's suggestion (below) for cleaning gummed up objectives.
...
From: RCHIOVETTI@aol.com
Date: Sat, 22 Sep 2001 14:22:32 EDT
Subject: Re: fouled objectives
Karl,
Whatever you do, please *don't* sonicate in acetone! This will dissolve the adhesives that seal the lens and keep the lens elements in place, and it will make the lens leaky. Acetone inside the lens will put you in deep trouble.
You can use anhydrous ether *very sparingly.* Use a cotton-tipped applicator or a "Q-Tip" moistened with
just a little ether and work quickly, gently using circular motions on the front lens element. The ether will
evaporate rapidly, and you don't want to rub the lens with a dry cotton applicator, so you will probably want
to use several applicators. Ether is good for this purpose because it evaporates so quickly. It's not around
long enough to damage the sealants and the adhesives.
You can follow this with a surfactant if necessary, but the ether should do the trick. Some surfactants can
leave a film on the surface of the lens unless you work quickly and follow with a rinse of distilled water
(using cotton applicators, *not* a stream from a squeeze bottle). I would avoid the use of surfactants if at all
possible. If you do need some surfactant action, first try a little commercial or consumer glass cleaner.
Check the labels for contents.
Remember, use ether sparingly, only enough to moisten the cotton applicator. Avoid flooding the front lens
element with ether (or alcohol, or anything, for that matter).
Also, please note the usual cautions when working with ether (open flames, sparks, electrical discharges,
etc. should be avoided, use adequate ventilation or work under a fume hood, etc.)
If you feel uncomfortable performing these steps, please call in a good microscope service technician who
knows what he/she is doing. I'm sure there is one in your area. This can save you a lot of headaches (and a
lot of lenses).
Good luck!
Bob Chiovetti
GTI Microsystems
Leica Exclusive Regional Dealer
Desert Southwest (Arizona, New Mexico, West Texas USA)
***Disclaimer: These are my own opinions, gained from experience, not
necessarily those of GTI Microsystems or Leica Microsystems.***
From: Monson, Frederick C. : fmonson@wcupa.edu
Date: Mon, 24 Sep 2001 09:09:23 -0400
Subject: RE: fouled objectives
Morning Karl,
My opinion only!
Since many/most of our best objectives/oculars are coated for color correction, Please don't use acetone on
an objective - EVER!!! Please don't use 100% ethanol, xylene or toluene on an objective - ever!? You may
concoct your own cleaner with non-ionic detergent (25%) and small amounts of alcohol(s) and (ONLY IF
REQUIRED) small - added when required - amounts of xylene or toluene. I have known optical technicians
who would rather scrape a lens with the edge of a new scalpel blade or a fragment/piece of double-edged
razor blade than expose a lens to any solvent for more than a brief moment for final - residual oil - removal.
There are some who never "rub". They "blow" with compressed air!
I have in my possession a product called "ROR" (URL: http://www.ror.net/ ) which smells like a regular
glass cleaner, but has received "KUDOS" from photographic folks for its effect on residual oils on lenses.
Thus, it appears that this is a product that does not remove color corrective coatings from photographic
lenses.
I have also had in my possession for many years a product I acquired from Xerox called: "Lens and Platen
Cleaner", Reorder No. 8R1025 (6 bottles) and "Cleaning Absorbent" [really clean cotton], Reorder No.
8R25. I have used this for residual oil removal for much longer than ROR, and I have never been
disappointed, though, of course, the Xerox cleaner was intended for the windows and optics in their copiers.
The biggest problems in cleaning microscope optics are these:
1. corrective coatings which wear the more they are cleaned,
2. mixing oils from different manufacturers (mentioned in a recent email to the listserver), because they can
react badly with one another and harden,
3. front surface mirrors which are usually inside fluorescent cubes or other complicated optical devices
(these should be cleaned only by someone who can afford to replace them),
4. permitting recidivist users to continue using better microscopes, and
5. assuming that a new faculty member or post doc has been properly taught or has properly learned while a
graduate student. (I will never forget the senior graduate student who removed an oil immersion lens
on a brand new microscope and poured it full of oil (from the nearby pint bottle) as he followed the printed
directions for its use!) I have NEVER kept a pint bottle near a microscope since!
To your specific, other, and possibly implied questions (more than
you ever hoped for in one man's opinion):
1. Yes, you can clean the 'dry' objectives of caked on oil with fluids. These are easier than many older oil
immersion objectives, because their exposed lens surfaces are usually flat.
2. Cleaning is preferable to discarding without trying.
3. Serious caked on oil can be scraped off (GENTLY!!!) before trying to finish cleaning with a solution.
4. No, please do not soak the lens in anything.
5. Please, do not subject a complicated multi-element lens system to immersion and ultrasound. You will
NOT 'see the baby' by doing this. The ultrasound will actually tend to separate cemented lens combinations
and to dissolve or fragment the cement that seals the face element to the surrounding metal jacket. NO,
please, definitely NO ultrasound for microscope objectives, oculars, mirrors, filters, or prisms.
I HAVE used ultrasound to help clean grease from microscope focusing mechanisms and from sticky leaf
diaphragms, but after you dismantle your first leaf diaphragm, you will know what real trouble is.
One way to help users is to predefine iris and field diaphragm adjustments for each objective and mark the
positions of those adjustments so that indices are easily repeated. This is far more important for the
iris diaphragm than the field, because most unschooled microscopists learn in their first class to use the iris
diaphragm for contrast enhancement (a.k.a., resolution diminution!). By placing the settings in plain view
you will give yourself the opportunity to review the purposes of the two diaphragms with each new user and
prevent unnecessary exposure of microscope internals.
Is it obvious that I have a fixation on these matters?
From: Leona Cohen-Gould : lcgould@med.cornell.edu
Date: Mon, 24 Sep 2001 14:07:49 -0400
Subject: Re: fouled objectives
Hi Karl,
I manage a multi-user optical microscopy facility. Brace yourself. You will have to resign yourself to the
fact that no matter how carefully you explain things to your users, someone is going to drag a dry lens
through oil, get fingerprints on the ocular lenses, and do things you hadn't imagined. I try (not always
successfully) to keep a schedule of visually inspecting the lenses, etc and correcting the damage before it
gets too far gone. both of our microscopes are inverted, so I have had to devise little "diapers" for the lenses
to absorb the excessive amount of immersion oil my users feel they need, so that it doesn't drip down and
work its way into the lenses or even the body of the 'scope itself! (yes, they are that sloppy).
We routinely use a solution of Windex (the standard blue stuff) & water (1:1) to keep our lenses clean. It
works very well on oil that is still liquid. For the encrusted lenses, you may need something stronger. I
would call your sales/service rep for the microscope and ask what they recommend for their lenses, since
the coating on the lenses may be damaged by different solvents.
Vigilance pays. Good luck.
Lee
--
Leona Cohen-Gould, M.S.
Sr. Staff Associate
Director, Electron Microscopy Core Facility
Manager, Optical Microscopy Core Facility
Joan & Sanford I. Weill Medical College
of Cornell University
New York, New York
From: Sara Miller :
Date: Thu, 27 Sep 2001 10:32:38 -0400 (EDT)
Subject: Re: fouled objectives: summary (fwd)
The EM supply houses carry special (clean & without grit) styrofoam sticks for cleaning diamond knives;
these are fairly inexpensive. I'm sure these would be a good source for scratch-free styrofoam with which to
clean lenses.
Sara E. Miller, Ph. D.
P. O. Box 3712
Duke University Medical Center
Durham, NC 27710
**
GM comments:
A popular surfactant is “Sparkle” a purple color glass lens cleaner available by the gallon in Chicago and by
the ounce from local microscope dealers.
At the end of the imaging session (and during if necessary) I routinely wipe oil off the (oil immersion)
objective lens by taking a single sheet of new lens paper, folding it breadth-wise several times (to make a
long skinny paper with greater strength) and then wiping one time along the length of the paper. Never rewipe
– doing so can put the lens at risk of scratches from dirt transferred to the lens on the first wipe.
If you “dunk” a dry objective lens in oil it suggests to me that either (a) you were working too fast or are
inattentive, or (b) you don’t know how to drive – if the latter, get out of the driver seat and have someone
who knows what they are doing operate the microscope for you. I admit to the former faults -on rare
occasion ☺ - when it happens to you, slow down and concentrate on using the instrument correctly. Lastly,
if you “dunk” a dry objective, or suspect that someone before you did (i.e. hazy 20x images), inform the
Core staff and we will clean the lens.
McBain Instruments (S. California Leica dealer) recommends Sparkle or methyl ethyl ketone (MEK),
sparingly, on a cotton tip, to clean aqueous or organic build up on objective lenses.
GM: to clean the viewing side of eyepieces, we “huff” on the lens to moisten the lens (condensation of
breath) followed by gentle wiping with clean fabric (shirt). We inspect the lens before doing this to make
sure that there are no particles on the lens that might act like sandpaper (remove such particles using a
cotton tip as described above for objective lenses.
**
Hi,
I was the microspectrophotometry specialist at Zeiss when one of our product managers
discovered Sparkle. We, also, had used ether up to that point (I still prefer it, when used
cautiously, with good ventilation). Since Zeiss is very conservative and would have tested
thoroughly before recommending such a drastic switch, I would have confidence in Sparkle.
I agree with Ian re: cleaning method, however, first gently daubing the excess oil off with a piece
of lens tissue. I then switch to pure cotton swabs (Q-Tips are OK... if another brand, make sure
that they are 100% cotton). Gently moisten just the tip; shake off any extra, then swirl from the
center to the outside rim. Repeat with a clean swab if necessary.
If you or your group use oil constantly, you can just clean off the excess each day then give a
thorough cleaning once a week. It will not hurt to leave a thin sheen of oil on the front surface of
the lens over that period. However, if oil is left, especially in the outer ridge where the lens meets
the housing, it can polymerize and harden. At that point cleaning is both difficult and hazardous.
The hardened polymer can chip away in fairly sharp shards and, if you are not careful, scratch the
anti-reflection coating on the surface of the lens.
One other tip: Since the front windows on high NA objectives are very small, sit your objective on
its screw end under a stereo for easier viewing. For a quick look, remove an eyepiece and flip it
upside down. Hold it at about a 45 degree angle from the surface you are inspecting; nearly
touching.
Hope this is helpful.
Barbara Foster
Microscopy/Microscopy Education
125 Paridon Street, Suite 102
Springfield, MA 01118
**
{too bad post below is not explicit on what "meths" is -- maybe, or not, methanol):
I've just switched from ether because of safety concerns. I investigated all
the alternatives but after speaking to a Zeiss engineer, of the old school, have
started using meths. Clean the excess oil from the objective with lens tissue then
using a fresh piece of lens tissue lightly damped with meths gently clean the
objective. So far, in my hands, it's been very successful.
Ian.
Dr. Ian Montgomery,
Histotechnology,
Graham Kerr Building,
Institute of Biomedical & Life Sciences,
University of Glasgow,
Glasgow,
G12 8QQ.
|
|
Cleaning oil immersion objective lenses advice from confocal listserv 2/2022 parts of thread (occasionally, i.e. monthly - usually done by core staff)
Subject:
Re: Oil objective lifetime
Date:
Fri, 11 Feb 2022 09:45:56 -0700
From: Craig Brideau
I'm a bit alarmed to hear what some of you are dealing with in terms of oil
films and solvents! I've just used isopropanol and a premium optical-grade
tissue wipe on all our lenses for the last 16 years and have encountered
none of these problems. I've been exclusively using Cargille oil, which I
realize may not meet the higher optical standards many of you are aiming
for, so it could be a difference in oil formulation. Just wanted to express
my surprise at how vexing this problem is for some.
Craig
On Fri, Feb 11, 2022 at 9:24 AM Steffen Dietzel <lists@sdietzel.de> wrote:
*****
Hi Dan,
just to clarify: Are you talking about Type F immersion oil from Zeiss,
the stuff that is still currently in use?
Type F-Immersion oil can oxidize? We use that exclusively and never
noticed anything weird when leaving a thin film on the objectives. So
far, I am more worried about users scratching the front lense during
'cleaning' rather than small amounts of oil remaining. Now you gave me
something to worry about.
Best
Steffen
---
11.02.2022 (Feb 11, 2022) dan@bioptechs.com:
*****
Oilers
I’ve been watching this thread and thought this comment would be
beneficial. When I was a much younger guy employed by a then Zeiss
Franchise in Md. covering the central east coast, I often came across
objectives that were only partially wiped off or partially cleaned.
This resulted in a thin layer of oil remaining on the objective that
would easily oxidize, especially overnight! This thin oxidized layer
had an irregular refractive index. Then when additional oil was added
for imaging you can imagine what happened to the image. I found the
best way to clean them was to, after removing the bulk of the visible
oil, use a strong solvent such as lighter fluid to dissolve the
thicker oxidized film followed by a water based cleaner. The petroleum
cut through the oil and oxidized film (stuck to the glass) and the
water based cleaner dissolved any remaining, or acquired salts and
sugars that my have accidentally deposited from media or handling. It
is important to note that when using a petroleum solvent you don’t
saturate the wiping tissue, only make it “damp” and limit the amount
of time exposure to the lens. Many times the user would be surprised
at how improved the image was. For wiping paper I strongly suggest
Berkshire, Lensx 90.
https://berkshire.com/shop/cleanroom-wipes/nonwoven/lensx-90/ln90040624p/
<
https://berkshire.com/shop/cleanroom-wipes/nonwoven/lensx-90/ln90040624p/>
It is the softest, most absorbent optical wipe I ever used. I have no
interest in the company but I haven't found anything better for lenses.
Dan - Dan Focht, Bioptechs
On Feb 11, 2022, at 6:09 AM, Sylvie Le Guyader wrote:
*****
Dear list
I add a couple of things to this discussion:
- we do not want to have petroleum in our facility as a routine so
our users are instructed to use ethanol to clean the objectives after
usage. We use the cleaning procedure shown in this excellent
video<https://www.youtube.com/watch?v=Tz4Dy5D6kdw> by Kurt Thorn. The
facility staff inspects all objectives on each microscope every 2
months and clean them with petroleum. This is because we realized the
risk in case of petroleum spill after doing a risk assessment. We are
well aware that ethanol has the wrong polarity and therefore it does
not fully clean the immersion oil as thoroughly as petroleum. This
means that the objectives have a light coating of immersion oil
pretty much all the time. Our oldest objectives are 16 years old. We
have never had any issue with the lens detaching. This said, the air
objectives are only in contact with oil for short periods of times
until we discover the issue and clean them.
- To avoid having the objective barrel coated in oil, we instruct our
users to wipe off the excess oil from the lens and the metal around
the lens with a dry lens paper (no ethanol) every time they change
dish or slide the add new oil for the next slide. Cleaning with
ethanol is only done at the end of their session. This has 2
advantages: it prevents the oil flattened by the objective to flow
further down the objective barrel with every slide. It also removes
potential air bubble in the oil which often show up when one removes
a sample.
Med vänlig hälsning / Best regards
Sylvie
Sylvie Le Guyader, PhD
Live Cell Imaging Facility Manager
Karolinska Institutet- Bionut Dpt
Hälsovägen 7C,
14157 Huddinge, Sweden
|
|
Microscope illumination stability testing (see web page for full content)
Illumination Power and illumination stability
Nathalie Gaudreault et al 2022 Illumination Power and illumination stability
https://www.protocols.io/view/illumination-power-and-illumination-stability-bzp8p5rw
DISCLAIMER:
This protocol was developed by the members of QUAREP-LiMi Working Group 1. The member list can be found here: (https://quarep.org/working-groups/wg-1-illumination-power/wg-1-members/)
The QUAREP-LiMi is a group of scientists interested in improving quality assessment (QA) and quality control (QC) in light microscopy. We first came together on April, 2020 and as of February 2022 the group has grown to 395 people from 34 countries spread around the world. We have members from academia, microscopy communities, companies, organizations or institutions related to standardization, scientific publishers, and observers from funding agencies.
ABSTRACT
To obtain accurate, reproducible, and interpretable data when conducting imaging experiments, it is critical to consider external factors affecting data acquisition at various steps of the experimental workflow. Illumination power and stability represent two critical factors, especially when comparing fluorescence intensities between images during a time-lapse experiment or experiments performed at different times or on other microscopes.
The fluorescence signal can be generated by different types of light sources. These light sources and their coupling elements (e.g., fibers) can display varying performances over time as they age, move, or as environmental conditions change.
Unfortunately, microscope users can often only set illumination power as a percentage of its maximal output and, may therefore, not be aware of potential performance changes. It is important to recognize that a set percentage will not always yield the same illumination power in Watts at the objective over the course of an experiment, not to mention between days or systems. This means that selecting for example 10% output may lead to different experimental results over time and will not necessarily be comparable to outputs obtained from other lasers or microscopes, even those of the very same model.
If you are responsible for system maintenance, routinely measuring the illumination power, stability, and linearity over time can help you detect issues that affect the integrity of the system and thus the reproducibility of an experiment.
This protocol describes how to measure the stability and linearity of the illumination power using calibrated external power sensors. This protocol is at the moment intended for confocal systems (raster scanning and spinning disks), but will be extended later to other imaging modalities. It represents the collective experience of 60 imaging scientists. Measurements made by our working group with this protocol are available in a public database, which will be updated with further contributions from the community.
==> see web page fore full content.
|
|
Excellent quote (applies to microscopy learning curves and more)
Lesson #3: Practice, practice, practice
In theory there is no difference between theory and practice. In practice there is. – Benjamin Brewster
found at https://www.pixelbiotech.com/post/what-we-learned-about-image-analysis-after-processing-thousands-of-customer-datasets
|
|
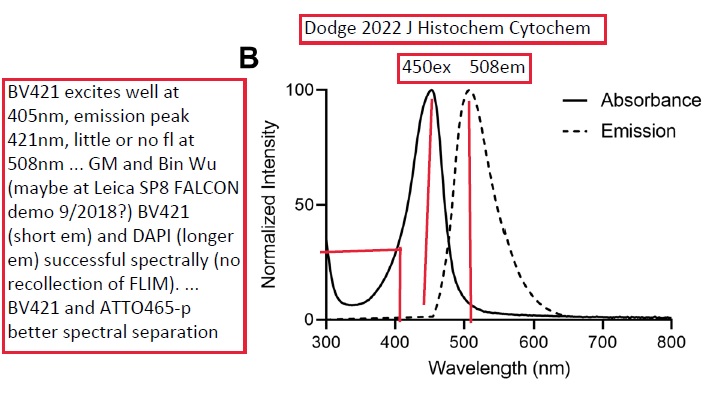
Potential future DNA counterstain - put more valuable fluorophores on 405nm excitation, such as BV421 and other Brilliant Violets.
Joshua T Dodge, Andrew D Doyle, Ana C Costa-da-Silva, Christopher T Hogden, Eva Mezey, Jacqueline W Mays
Atto 465 Derivative Is a Nuclear Stain with Unique Excitation and Emission Spectra Useful for Multiplex Immunofluorescence Histochemistry
J Histochem Cytochem. 2022 Mar;70(3):211-223. doi: 10.1369/00221554211064942.
PMID: 34994225 DOI: 10.1369/00221554211064942
https://journals.sagepub.com/doi/10.1369/00221554211064942
Multiplex immunofluorescence (mIF) is an effective technique for the maximal visualization of multiple target proteins in situ. This powerful tool is mainly limited by the spectral overlap of the currently available synthetic fluorescent dyes. The fluorescence excitation wavelengths ranging between 405 and 488 nm are rarely used in mIF imaging and serve as a logical additional slot for a fluorescent probe. In the present study, we demonstrate that the addition of 2,3,4,5,6-pentafluoroaniline to Atto 465 NHS ester, creating Atto 465-pentafluoroaniline (Atto 465-p), generates a bright nuclear stain in the violet-blue region of the visible spectrum. This allows the 405 nm excitation and emission, classically used for nuclear counterstains, to be used for the detection of another target protein. This increases the flexibility of the mIF panel and, with appropriate staining and microscopy, enables the quantitative analysis of at least six targets in one tissue section.
Keywords: Atto 465; SP8; immunofluorescence; immunohistochemistry; multiplex; nuclear stain; photoconversion; photostability; proflavine; tyramide.
|
|
colocalization - 3D data vs Maximum Intensity Projection (MIP)
Assumption: trying to colocalize molecules in one cell (alt: if just seeing if same cell expresses two things, MIP ok if single cell layer).
GM simple advice:
1. Use 3D with microscope at "best available resolution", ex. 1.4NA objective lens, 50 nm XY pixel size, 150nm Z-step size, pinhole 1.0 Airy unit (for each channel), bright fluorophores (how about Brilliant Violet BV421 and Brilliant Blue BB515?!).
2. if you have access to a super-resolution microscope (aka nanoscope), use it - if you would like to by our core a STED (stimulated emission depletion) and/or other nanoscope, we welcome it (we can find space in core - hopefully you can also contribute ongoing service contract costs and part or all of core manager's salary, overhead, etc).
Simple thought experiment -- suggest consider two cases: very flat cell (ex COS-7 or U2OS) vs spherical cell (resting T-cell, B-cell) ...
live cell labeling fluorescent wheat germ agglutinin of plasma membrane + Hoechst and/or PicoGreen DNA counterstains ... MIP would show plasma membrane and nuclei colocalized, optional PicoGreen mito nucleoids.
***
nice example (GM annotations of figure 2 - also cropped out panel "iii" to save space):
My go-to illustration of the unsuitability of MIPs, and the effect of cell geometry and anisotropic voxels, in colocalization analysis is figure 2 in this article from 2008:
https://pubmed.ncbi.nlm.nih.gov/18353895
Chris Wood
Laboratorio Nacional de Microscopía Avanzada-UNAM
Instituto de Biotecnología
Universidad Nacional Autónoma de México
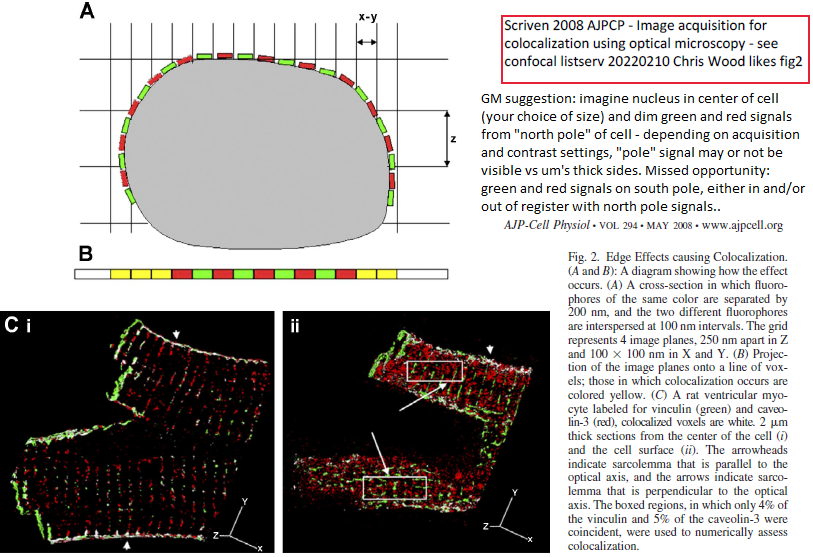
***
Confocal listserv exchange February 2022 - more content in message thread online
Subject: Re: Colocalization MIPs versus 3D Volume
Date: Wed, 9 Feb 2022 09:07:31 +0000
From: Jeremy Adler <jeremy.adler@IGP.UU.SE> [Jeremy's email here because readily available from publications]
Reply-To: Confocal Microscopy List <CONFOCALMICROSCOPY@LISTS.UMN.EDU>
To: CONFOCALMICROSCOPY@LISTS.UMN.EDU
*****
Hej Kathy,
Quantitfying colocalization is a minefield - MIP is clearly nonsense and will change as the thickness of the Z series increases.
1) there are too many colocalization coefficients and some are almost meaningless.
In a recent article we make a case for abandoning one group of coefficients.
The premise is that colocalization coefficients fall into two useful groups, those that measure co-occurrence, the degree to which molecules are found in the same place.
And correlation, the intensity relationship when fluorophores are found together. Both measures are informative.
However this scheme exposes a third group that combine the two types of measurement into an unintelligible mess - these we propose dumping as the measurement can arise from widely differing distributions.
.
Adler, J, Parmryd, I.
Quantifying colocalization: The case for discarding the Manders overlap coefficient. Cytometry. 2021; 99: 910- 920.
https://doi-org.ezproxy.its.uu.se/10.1002/cyto.a.24336
.
2) A second serious problem arises with point scanning images, noise - two images taken consecutively of the same fluorophore are not measured as being perfectly correlated.
So if two nominally identical images don't correlate perfectly then correlations from two different fluorophores will not be measured correctly.
We demonstrate a practicable solution.
J. Adler, S.N. Pagakis and I. Parmryd (2008)
Replicate Based Noise Corrected Correlation for Accurate Measurements of Colocalization
J. Microscopy 230(1),121-133.
.
3) Expressed proteins with a fluorophores can cause problems, colocalization can be measured accurately but the levels of expressed protein are usually superimposed the endogenous protein, and will distribute differently - binding sites are saturable.
.
Overall there are some serious problems to resolve - segmentation is another.
-----Original Message-----
From: Confocal Microscopy List <CONFOCALMICROSCOPY@LISTS.UMN.EDU> On Behalf Of Kathryn Spencer
Sent: Tuesday, February 8, 2022 8:56 PM
To: CONFOCALMICROSCOPY@LISTS.UMN.EDU
Subject: Colocalization MIPs versus 3D Volume
*****
To join or leave the confocal microscopy listserv or to change your email address, go to:
https://lists.umn.edu/cgi-bin/wa?SUBED1=confocalmicroscopy&A=1
Post images on http://www.imgur.com and include the link in your posting.
*****
Hi everyone;
I'm looking for good analogies, tutorials, lessons, graphics, ideas, etc to convince our users that they should be using our Imaris license to determine colocalization in confocal Z-stacks instead of maximum intensity projections in FIJI. Since they see 'differences' in treated versus untreated in their MIP colocalizations analyses, they are happy. I'm pulling my hair out that their analyses are not done properly, and we have the super nice software that can handle 3D data sets.
Do you have any good resources, publications, or data sets to share to show why they should NOT be using MIPs to quantitate colocalization in confocal Z-stacks? My hand-waving representations and hand-drawn artistic abilities don't seem to be convincing enough.
I'm trying to obtain their data sets to analyze different ways, but would you have more simple lessons?
Thanks so much.
Best;
Kathy
Kathryn S. R. Spencer, Ph.D.
The Scripps Research Institute
Core Microscopy
|
|
https://blog.addgene.org/antibodies-101-buffers-storage-and-conjugates
Antibodies 101: Buffers, Storage, and Conjugates
By Rachel Leeson
Feb 1, 2022
Storage, buffers, conjugates ... bottom of page "More resources" = more antibody 101 pages.
|
|
D2O, Oxyrase, more ways to improve photostability of fluorophores: Kwon 2022 review mentions both.
D2O: see also Maillard 2021 Chem Sci 12, 1352 (see alsewhere on web site for details).
Oxyrase: GM has been a fan of Oxyrase (www.oxyrase.com) since learning about it from Clare Waterman (now a senior investigator at NIH) who learned about it from Gregg Gunderson's lab (GG was a MetaMorph customer). Until this review GM was unaware of key role of lactate in this product (guessed it would be glucose oxidase and catalase ... which perhaps one or both could be beneficial or maybe already inside).
Below from: Kwon 2022 AdvSci - SRM photobleaching off-states Bleaching‐Resistant Super‐Resolution Fluorescence Microscopy - photodestruction photostabilization.pdf DOI: 10.1002/advs.202101817
For multicolor SRM,different fluorophores often undergo distinct photoswitching mechanism and therefore each requires specific buffer compositions that limit fluorophore combination. For example, cyanine dyes operate optimally in oxygen-free conditions but rhodamine dyes do not blink well in the absence of oxygen.[47] Oxyrase-based anaerobic conditions (OxEA) were proposed as global conditions for fluorophores because it supports only small amounts of molecular oxygen.[70,71] Oxyrase is a sterile solution of membrane fragments from Enterococcus coli that specifically contain enzymes that catalytically convert molecular oxygen.[157] OxEA requires DL-lactate as a substrate and supports 1–2% of the steady-statemolecular oxygen concentration without significantly changing the solution pH, as well as the intracellular functions allowing live-cell imaging.[158]. As a result, OxEA permits efficient photoswitching of both cyanine and rhodamine fluorophores under the same buffer compositions, enabling simultaneous multicolor GSDIM imaging with Alexa488, Alexa555, and Alexa647 (Figure 2J,K).[71]
Alternative photostabilizing buffers include the sulfite buffer and the heavy water (D2O) instead of normal water (H2O). [72-75]
Oxyrase references:
[70] L. Nahidiazar,M. Kreft, B. van den Broek, P. Secades, E.M.M. Manders, A. Sonnenberg, K. Jalink, J. Cell Sci. 2015, 128, 3714.
[71] L. Nahidiazar, A. V. Agronskaia, J. Broertjes, B. van den Broek, K. Jalink, PLoS One 2016, 11, 0158884.
[157] K.-C. Ho, J. K. Leach, K. Eley, R. B. Mikkelsen, P.-S. Lin, Am. J. Clin. Oncol. 2003, 26, 86.
[158] T. Gudzenko, C. M. Franz, Front. Mol. Biosci. 2020, 7, 149.
D2O references (which in turn cite Stryer and other):
[72] T. M. P. Hartwich, K. K. H. Chung, L. Schroeder, J. Bewersdorf, C. Soeller, D. Baddeley, (Preprint) BioRxiv:465492, 2018.
[73] S. F. Lee, Q. Vérolet, A. Fürstenberg, Angew. Chem., Int. Ed. 2013 52, 8948.
[74] K. Klehs, C. Spahn, U. Endesfelder, S. F. Lee, A. Fürstenberg, M. Heilemann, ChemPhysChem 2014, 15, 637.
[75] W. Q. Ong, Y. R. Citron, J. Schnitzbauer, D. Kamiyama, B. Huang, Chem. Commun. 2015, 51, 13451.
First author is currently (1/2022) at JHU: Jiwoong Kwon is currently a postdoctoral research associate at Johns Hopkins University. ... research interest includes the development and applications of super-resolution imaging, and his current research focuses on DNA damage response and translation dynamics.
|
|
Double sided tape as channel walls of specified height
We use 3M double sided tapes of known thickness as spacers between 2 coverslips: 9415 (80um) and 467MP (50um) and match the refraction indices of the objective immersion medium and the sample.
Subject: Re: Calibration slide for focus height
Date: Sat, 22 Jan 2022 10:14:16 +0000
From: Sylvie Le Guyader - confocal microscopy listserv
GM note: 3M = Scotch tape. Double sided scotch tape also useful to put on back side of slide to make easier to pick up from inverted microscope holder -- especially our Olympus FV3000RS confocal microscope's OkoLab slide holder, that lacks 'finger holes" to reach around slide.
|



